Indice
Anterior
Siguiente
Correo Científico Médico de Holguín 2004;8(2)
Presentación de caso
Policlínica Comunitaria Docente “Julio Grave de Peralta” Holguín
Síndrome de Wiskott Aldrich. Presentación de un caso.
Wiskott Aldrich Syndrome. One case presentation
Mario Martín Jimenez1, Ondina Arias Figueiras2.
1 Especialista de primer
grado en Pediatría. Instructor de Pediatría. Policlínica “Julio Grave de Peralta”.
Holguín
2 Especialista de primer grado de Medicina General Integral. Policlínica
“Julio Grave de Peralta”. Holguín. Máster en asesoramiento genético.
RESUMEN
Se presenta un caso con el síndrome de Wiskott Aldrich, inmunodeficiencia congénita ligada al sexo que requiere para su diagnóstico, criterios clínicos – genéticos, hematológica e inmunológicas; se exponen datos clínicos y resultados de exámenes complementarios. Se revisa la literatura médica relacionada con dicha entidad.
Palabras clave: Inmunodeficiencia congénita ligada al cromosoma X/Diagnóstico.
ABSTRACT
A case of Wiskott Aldrich sindrome, congenital inmunodeficiency associate with X cromosome. Requiring clinical and hemathological criteria for its diagnosis is reported clinical data, complementary examinations are also reported. The medical literature regarding such entry is reviewed.
Key words: Congelital inmunodeficiency associate with X cromosome/diagnosis.
INTRODUCCIÓN
Las enfermedades por inmunodeficiencias heredadas son poco comunes, pero están relacionadas con severas patologías y elevada mortalidad. Pueden presentarse como una anormalidad aislada o asociada a otros defectos.
En las inmunodeficiencias primarias la alteración puede ser consecuencia de la afectación de la inmunidad celular relacionada con un anormal desarrollo de los linfocitos T específicamente o estar provocados por un dańo en el sistema humoral, donde el problema está directamente relacionado con un defecto en los anticuerpos. En otras ocasiones pueden clasificarse como mixtas, existiendo una alteración de ambos sistemas. Las mismas pueden ser consecuencia de la modificación en la cantidad y/o calidad de los elementos que intervienen en la respuesta inmune.
El síndrome de Wiskott Aldrich es un trastorno recesivo ligado al cromosoma X que implica deficiencias de las plaquetas y de las células B y T. Se debe a mutaciones en el gen WAS (Wiskott Aldrich Sindrome) situado en el brazo largo del cromosoma X exactamente en la banda 11.22 - 11.23 cuyo producto proteico es necesario para la normal formación del citoesqueleto y se plantea además que es un regulador en la función de linfocitos y plaquetas.
En los estudios inmunológicos el patrón predominante es una IgM sérica baja, IgA e IGE elevadas y una IgG normal o discretamente baja y sorprende que la subclase IgG2 es normal.
Por tratarse de una enfermedad poco común con una incidencia de dos casos en nuestro municipio pero con gran connotación familiar y social, nos sentimos motivados a presentar el caso con el propósito de profundizar en su conocimiento en cuanto a las características clínicas y hematológicas de la enfermedad.
MÉTODO
Se realizó la presentación del caso con ayuda de la Historia Personal o Ambulatoria, familiar y hospitalaria. Los datos además fueron recogidos mediante interrogatorio, el examen físico y los complementarios realizados y resultados de estudios del Instituto Nacional de Hematología. Se confeccionó el árbol genealógico familiar que facilitó ilustrar el patrón de herencia de la enfermedad.
Se realizó la evaluación clínico – genética, hematológica e inmunológica del caso, además de una revisión bibliográfica actualizada sobre el tema.
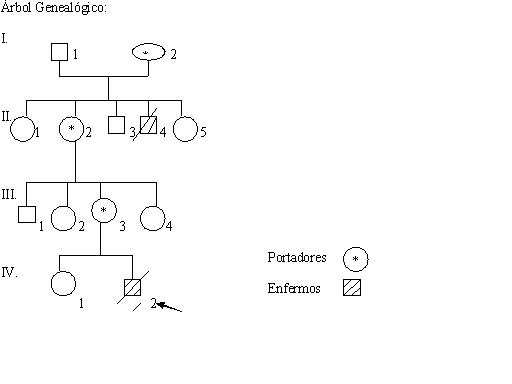
DESARROLLO
Presentación del caso:
Paciente masculino que ingresa a los 53 días de nacido por un cuadro febril; inespecíficos asociado a cifras bajas de Hb de 64 g/l, deposiciones oscuras, hepatomegalia, esplenomegalia, medulograma normal, plaquetopenia en 4 x 109/l, reticulocitosis en 60 x 103, se interpreta como una anemia hemolítica para estudio, al cuadro se unen infecciones respiratorias bajas de etiología bacterianas y cuadro dermatológico vesículopapuloso en tórax y abdomen, el paciente evoluciona tórpidamente presentando una bronconeumonía bacteriana y cuadro petequial en extremidades y tórax que requiere sun ingreso en UTIP, el cuadro dermatológico se modifica apareciendo lesiones escamosas en cuero cabelludo, cuello y región paraesternal compatible con una dermatitis seborreica planteándose la sospecha de una histiocitosis. Después de 3 ˝ meses de estadía hospitalaria se remite al Instituto de Hematología, Inmunología, regresa al mes con igual cuadro clínico y hematológico y con test de Isohemaglutinina que cuantificó de forma indirecta las cifras relativas de IgM que resultaron bajas.
Existiendo el antecedente patológico familiar de un tío materno fallecido en edades tempranas de la vida con cuadros respiratorios, sangramientos nasales y erupciones en la piel.
Se planteó el diagnóstico del Síndrome de Wiskott Aldrich y se solicita completar estudios de HLA a la hermana para valorar un posible trasplante de médula ósea.
A los 7 meses de edad reingresa en la UTIM con 11 días de estadía por procesos respiratorios virales y cuadros hematológico igual al descrito ya.
A los 7 ˝ meses requiere de ingreso en UTIP con cuadro de sepsis, Hb en 50 g/l, gasometría con PCO2 baja, plaquetopenia en 2 x 109/l, leuco 17 x 109/l y cuadro febril, en las primeras 24 horas desarrolla toma motora dada por monoparecia braquial derecha, presenta una convulsión tónico clónica generalizada, empeora progresivamente y fallece a las 48 horas de estadía por hemorragia intracraneal post infecciosa.
DISCUSIÓN
El síndrome de Wiskott Aldrich, es una patología infrecuente en nuestro medio, de carácter herediario, con un patrón autosómico recesivo ligado al sexo, donde solo las personas del sexo masculino la padecen. Es curable en algunos casos por medio de trasplante de médula ósea o, linfocitos T.
Se caracteriza clínicamente por presencia de dermatitis atópica, púrpura trombocitopénica con megacariocitos de aspecto normal pero con plaquetas defectuosas unido a una susceptibilidad insólita a las infecciones con frecuentes y prolongadas hemorragias y diarreas sanguinolentas.
Se invoca que la causa de la trombocitopenia parece ser una anomalía plaquetaria intrínseca, donde la supervivencia de las plaquetas alógenas marcadas con CR-5 es normal pero no así las isogénicas.
Las infecciones recurrentes suelen presentarse en el primer ańo de vida y son generalmente provocadas por neumococos y otras bacterias con polisacáridos capsulares, dando lugar a otitis media, neumonías, meningitis, sepsis generalizadas, aumenta la frecuencia de infecciones por neumocistis carinii y herpes virus por lo que es raro que los nińos superen la adolescencia, siendo las principales causas de muerte las infecciones, las hemorragias y las neoplasias malignas asociadas.
Recientemente se aisló en gen SWA en el brazo corto del cromosoma X en XP 11.22 – 11.23 cerca del centrómero que codifica la proteína PSWA (Proteína del síndrome Wiskott Aldrich) necesaria para la normal formación del citoesqueleto y que además actúa como reguladora importante de la función de linfocitos y plaquetas.
En la literatura revisada se seńala también que existen descritos otros síndromes que se acompańan de trombocitopenias hereditarias con patrones de trasmisión autosómico recesivo, otros con patrones recesivos ligados al X, etc; lo que permite en algunos pacientes con púrpuras trombocitopénicas y ausencia de otros estigmas el diagnóstico de una variante del síndrome de Wiskott Aldrich como la Histiocitosis de Letter Siwe y el síndrome de Kasabach Merritt.
Con la identificación de los genes responsables de las inmunodeficiencias ligadas al cromosoma X, unido a la disponibilidad de los marcadores de ADN ligados permitió realizar pruebas o test a portadores mediante el estudio del patrón de desactivación del cromosoma X en linfocitos de mujeres con riesgo, aunque actualmente esta técnica está siendo suplantada por la iderntificación directa de las mutaciones de los genes responsables.
CONCLUSIONES
Se trata de una enfermedad infrecuente que puede resultar mortal y requiere de un manejo oportuno y conocimiento adecuado.
El diagnóstico necesita de criterios clínicos, genéticos, hematológicos e inmunológicos.
BIBLIOGRAFÍA