Hospital Docente Provincial “Vladimir Ilich Lenin”
Presentación de un caso de enfermedad de Von Recklinghausen.
Von Recklinghausen´s Disease. A Case Report.
Luis Gómez Peńa1, Ivonne Roche Cisneros2, Daimi Díaz Delgado3.
1 Especialista de Primer Grado en Medicina Interna. Profesor Asistente. Centro de Trabajo: Hospital Provincial Docente V. I. Lenin.
2 Licenciada en Enfermería. Centro de Trabajo: Policlínica Docente “Manuel Díaz Legra”.
3 Especialista de Primer Grado en Medicina General Integral. Profesor Instructor. Centro de Trabajo: Escuela Latinoamericana de América Latina.
RESUMEN
La neurofibromatosis 1 es una enfermedad genética, de herencia autosómica dominante, de alta penetrancia y expresividad muy variable. Se realizó una presentación de caso, seleccionando a un adulto de 27 ańos, de la raza negra, de procedencia haitiana, que desde nińo presentaba verrugas diseminadas en todo el cuerpo y en esta ocasión fue admitido en el Hospital L’ Providence, pues tenía un neurofibroma de gran tamańo que le imposibilitaba el decúbito supino, apenas dormía por las molestias que le ocasionaba y además en el orden estético le producía algunas dificultades. Se indicó tratamiento quirúrgico al neurofibroma gigante que presentaba. Constituyeron las lesiones óseas, las manchas café con leche, el retraso pondoestatural y el retardo mental ligero, las características clínicas más frecuentes vistas. Se concluye entonces planteando que la neurofibromatosis tipo 1 es una enfermedad que afecta esencialmente el crecimiento celular del tejido neural, su curso es progresivo a lo largo de la vida, el diagnóstico es eminentemente clínico, donde las lesiones cutáneas constituyen el elemento característico.
Palabras clave: neurofibromatosis, Von Recklinghausen, diagnóstico clínico.
ABSTRACT
The neurofibromatosis 1 is a genetic illness, of dominant autosomal heredity, high penetrance and very variable. A study was carried out, with a 27- year-old adult, black race, from Haiti .The patient had disseminated warts in the whole body since his childhood and he was admitted at L' Providence Hospital due to a neurofibroma of a great size that disabled him the supine,dorsal decubitus position, he hardly slept due to the difficulties that it caused him, even in the aesthetic order . Surgical treatment was indicated . The most frequent clinical characteristics were the bone lesions, café au lait spot, weight-height retardation and the slight mental retardation. The neurofibromatosis type 1 is a progressive illness that affects essentially the cellular growth of the neural tissue, the diagnosis is eminently clinical, where the cutaneous lesions constitute the characteristic element.
Key words: neurofibromatosis, Von Recklinghausen, clinical diagnosis.
INTRODUCCIÓN
Los síndromes neurocutáneos constituyen un grupo de afecciones hereditarias que comprometen siempre la piel y el sistema nervioso central (ambos derivados del ectodermo) y otros sistemas y aparatos, en forma y extensión extremadamente disímiles (1,2,3) La neurofibromatosis (NF) es una enfermedad genética progresiva con expresividad muy variable. Sus manifestaciones más frecuentes son cutáneas, neurológicas, oftalmológicas y esqueléticas, con alta incidencia de neoplasias, especialmente en el sistema nervioso central. (4)
La neurofibromatosis 1 (NF1) es una enfermedad genética, de herencia autosómica dominante, de alta penetrancia y expresividad muy variable. El 50% de los casos se debe a mutaciones espontáneas. El gen de la NF1 se encuentra en el brazo largo del cromosoma 17 (17q11.2) En 1987 se realizó el mapeo y en 1990, la clonación. El producto del gen de la NF1, denominado neurofibromina, contiene 2.818 aminoácidos con una masa molecular estimada en 220 kDa; está involucrado en el crecimiento y la diferenciación celulares, especialmente en el tejido nervioso.
Una porción codificada del gen NF1, de alrededor de un 13%, muestra una estrecha similitud con la proteína activadora de guanosín trifosfatasa (GAPasa) Las proteínas GAP intervienen en la regulación negativa de las proteínas activadoras de los protooncogenes ras. El gen NF1 tendría, entonces, una función supresora tumoral. Reafirma esta hipótesis la pérdida de heterocigocidad hallada en varios de los tumores que se manifiestan en tal afección. (5-7)
La histopatología de esta enfermedad encuentra neurofibromas cutáneos como lesiones circunscriptas, no encapsuladas, compuestas por fibras colágenas onduladas, delgadas y algo eosinofílicas, dispuestas en bandas laxas que se extienden en distintas direcciones; se observan entre ellas abundantes núcleos ovalados o fusiformes, de tamańo uniforme, ocasionalmente ubicados en hileras paralelas. La ausencia del soporte colágeno dérmico normal explica el orificio palpable en la piel.
Los fibroblastos endoneurales tienen tendencia a formar empalizadas y presentan cilindros entre ellos; hay acumulación de colágeno en el interior del tumor. Se observa gran cantidad de mastocitos, que pueden llegar al 5% del total de la población celular. Algunas veces, en parte del tumor o en su totalidad, se advierte degeneración mucoide del estroma. Los neurofibromas plexiformes involucran a nervios profundos, grandes y muestran fascículos irregulares como consecuencia del incremento de la matriz endoneural y del perineuro, sin aumento del número de fibras nerviosas. (8,9,10) Tiene como objetivo entonces esta presentación de casos, profundizar en algunos elementos clínicos y terapéuticos (quirúrgicos) de esta entidad poco frecuente y observada en un país subdesarrollado, donde fue posible realizar acciones de salud en este tipo de paciente, esta fue nuestra mayor motivación al realizar el estudio.
PRESENTACION DEL CASO
Se trata de un paciente de 27 ańos de edad con antecedentes de padecer de lesiones en la piel desde los seis meses de edad, en forma de verrugas que a medida que pasaron los ańos, aparecieron mayores cantidades de las mismas. En esta ocasión ingresó en el Servicio de Medicina Interna del Hospital L’ Providence, Gonaive, Haití, por presentar una tumoración de gran tamańo (neurofibroma plexiforme) en la región dorso lumbar izquierda que le imposibilitaba mantener el decúbito supino, molestaba mucho para dormir y además presentaba una zona desprovista de piel, expuesta, con abundante tejido de granulación, no infectada.
| Fotografía 1 |
 |
| Neurofibroma plexiforme |
Al examen físico del paciente se encontraron algunos elementos que ayudaron a establecer el diagnóstico de la enfermedad:
- Retraso en el desarrollo pondoestatural.
- Lesiones en piel tipo café con leche, que asentaban en los propios neurofibromas cutáneos y eran un tanto mas clara que el color propio de la piel de la raza negra.
- Neurofibromas de diferentes dimensiones, que aumentaron con la edad en tamańo y cantidad, diseminados en todo el cuerpo.
- Retraso mental ligero.
- Lesiones óseas características, presentes sobre todo en columna vertebral y en huesos largos de las piernas.
- Familiar de primer orden con similares lesiones cutáneas.
El diagnóstico de esta enfermedad se tuvo en cuenta según los criterios expresados en la clasificación emitida por la Revista Arch. Argent. Pediatr. 2003, el Instituto Nacional de la Salud de EE.UU. que los definió en 1987 y la NIH Consensus Developmernt Conference de 1988 que tuvo iguales consideraciones diagnósticas (11-14) donde se plantea que para el diagnóstico de neurofibromatosis tipo I, un paciente debe tener al menos dos de los siguientes criterios:
Tabla 1: Criterios diagnósticos de la neurofibromatosis tipo I, utilizados en este caso.
|
Criterios diagnósticos. |
|
Seis manchas café con leche o más, de más de 0,5 cm. de diámetro en prepúberes, y de más de 1,5 cm. en adultos. |
|
Dos neurofibromas o un neurofibroma plexiforme o más. |
|
Efelidoides axilares o inguinales. |
|
Lesión ósea distintiva, como displasia del esfenoides, adelgazamiento de los huesos largos, con pseudo artrosis o sin ella. |
|
Glioma óptico |
|
Dos nódulos de Lisch o más. |
|
Un familiar de primer grado con diagnóstico de neurofibromatosis. |
Fuente: Arch. Argent. Pediatr 2003
En nuestro caso estaban presentes cuatro de los siete criterios antes mencionados, las lesiones tipo manchas de café con leche en numero superior a seis, los neurofibromas cutáneos y uno plexiforme, las lesiones óseas distintivas de esta enfermedad y la presencia de la enfermedad en un familiar de primer orden.
A continuación enumeramos algunas imágenes que apoyan nuestro pensamiento diagnóstico:
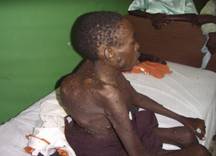
| Fotografía 2 | Fotografía 3 |
 |
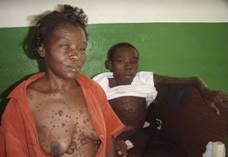 |
| Lesión ósea característica | Madre con neurofibromas |
| Fotografía 4 |
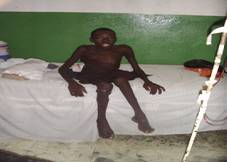 |
| Lesiones óseas de las piernas |
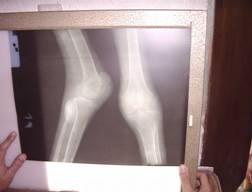
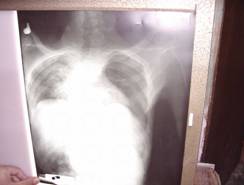
El diagnóstico es apoyado con estudios imaginológicos.
| Fotografía 5 | Fotografía 6 | |
 |
 |
|
| Neurofibromas en huesos de extremidades inferiores. | Lesión en columna vertebral |
El estudio histológico del neurofibroma resecado no se realizó por encontrarnos en un país del tercer mundo donde esta posibilidad no es posible por lo costoso del mismo.
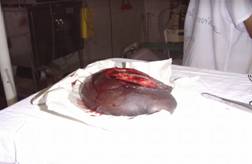
El tratamiento consistió en la extirpación quirúrgica del gran neurofibroma motivo de consulta del caso.
| Fotografía 7 | Fotografía 8 | |
 |
 |
|
| Neurofibroma gigante (plexiforme) | Piel después de resecado el tumor |
DISCUSIÓN
Durante el cumplimiento de la misión internacionalista en la hermana República de Haití, fueron innumerables los casos clínicos interesantes que sólo los libros lo muestran en nuestra patria. El caso que nos ocupó es un paciente que, clínicamente y basado en los criterios diagnósticos más utilizados al respecto, definimos se trataba de una neurofibromatosis tipo I, entidad que anteriormente se denominaba enfermedad de Von Recklinghausen.
Si tenemos en cuenta el motivo de consulta de nuestro caso, seńalaríamos que constituyeron los neurofibromas el sello distintivo en este paciente y sobre todo uno gigante (plexiforme) que era el que más dificultades en el orden estético le producía, pero además doloroso al adoptar ciertas posiciones en el decúbito e incluso en la posición de pie.
La literatura hace mención a tres tipos de neurofibromas, los cutáneos, los subcutáneos y los plexiformes. Los mismos aumentan en número y tamańo a lo largo de la infancia, sobretodo durante la pubertad, siendo junto al glioma óptico, la macrocefalia y el retraso del desarrollo pondoestatural los motivos más frecuentes de consulta (1) Los neurofibromas cutáneos que son los más frecuentes en este paciente se plantean que están situados en la dermis, son de consistencia blanda o firmes adheridos a la piel, de tamańo variado, así como sus formas aplanadas, sésiles, pediculados, lobulados. Cuando se comprimen tienden a invaginarse a través de un pequeńo orificio de la piel (signo de ojal) y la cantidad de los tumores puede variar desde pocos a miles (15), es visto en el 44% de los casos que reporta este artículo.
Estos resultados coinciden con los encontrados en nuestro caso. En relación con el neurofibroma gigante frecuente en el 57% de los casos vistos según el articulo que hacemos mención, se plantea que cuando crece de un tamańo considerable, a la palpación de los mismos hay una sensación de tocar “un saco de gusanos” y estos son los denominados neurofibromas plexiformes, característica clínica de igual similitud que lo observado en nuestro paciente.
Se conoce que estos neurofibromas alcanzan un tamańo enorme comprometiendo la piel, la fascie y puede llegar a incluir el músculo, infiltrar vísceras, erosionar estructuras óseas y que son más frecuentes en la región craneofacial, orofaringe, retrofaringe, región cervical y mediastino superior (1), no coincidiendo con nuestro paciente donde el neurofibroma plexiforme estaba localizado en la región dorso lumbar izquierda.
Las manchas café con leche observadas en neurofibromas son vistas según la literatura en el 9% de los casos (15), este dato fue también observado en nuestro paciente. Farreras Rozman plantea que estas lesiones constituyen junto a los neurofibromas el rasgo fenotípico característico de estas personas con una frecuencia de 1 de cada 3000 personas. (16)
Las lesiones óseas erosivas pueden dar lugar a cifosis, lordosis, luxaciones y fracturas frecuentes (15), el caso que nos ocupa presentaba una cifoescoliosis importante y lesiones óseas en huesos de las piernas que le imposibilitaba deambular (fotografía 3 – 6) En la decimocuarta edición del Farrearas Rozman (16) y en un artículo publicado en la ciudad de La Habana (17), se plantea que la escoliosis es más frecuente a partir de la primera década, tal como se presentó en este paciente. Seńala además que en las radiografías de los huesos largos aparecen destrucciones secundarias por los neurofibromas plexiformes o existen lesiones líticas circunscritas sin relación a los neurofibromas, aunque no realizamos radiografías en columna vertebral lumbosacra, si apreciamos en la de los huesos largos, la presencia de neurofibromas que producen compresión sobre el tejido óseo y que ha imposibilitado la marcha en este caso junto a la posible lesión vertebral dorso lumbar que pudiera existir. Enrique Galin Gómez (18) plantea que algunos pacientes tienen un arqueamiento tibial por seudoartosis lateral, este dato no fue visto en el paciente que nos ocupa.
En esta enfermedad existe una gran variabilidad clínica, pues existe una expresividad genética muy variable, por lo que pacientes incluso de una misma familia se manifiestan de diferentes maneras (18, 19), esto coincide con nuestro paciente si tenemos presente que su madre, de 49 ańos sólo manifiesta clínicamente las lesiones tipo neurofibromas cutáneos (fotografía 2).
Aunque existe riesgo de malignización de los neurofibromas como complicación, constituyen otras las más frecuentes tales como el retraso mental, la estatura corta y los cambios vasculares y neurológicos no relacionados con los neurofibromas (18), en nuestro paciente fueron vistos el retraso mental ligero y la estatura corta, además de los signos clínicos antes mencionados.
En caso de tumores gigantes o compromiso de estructuras importantes de nuestro organismo, se plantea que el tratamiento quirúrgico es la única opción. (15,19) Coincidimos con la literatura y esta constituyó la opción utilizada en el caso del neurofibroma plexiforme presente en nuestro caso.
Se concluye entonces planteando que la neurofibromatosis tipo 1 es un desorden genético que afecta esencialmente el crecimiento celular del tejido neural, su curso es progresivo a lo largo de la vida. El diagnóstico es eminentemente clínico, basado en los criterios antes seńalados, donde las lesiones cutáneas constituyeron el elemento característico.
BIBLIOGRAFÍA