Índice Siguiente
Correo Científico Médico de Holguín 2010;14(2)
Presentación de caso
Hospital
Pediátrico Universitario Octavio de
Servicio
de Imagenología. Holguín
Presentación de un paciente con síndrome de Crouzon
Presentation
of a Patient with Crouzon’s Syndrome
Janice
Morales Rodriguez1, Martha Sarmiento
Oliveros2,
Melba Zaldivar Santiesteban3, Miguel Leyva Tamayo4, Tania
Garnier Avila5
1
Especialista de Primer
Grado en Imagenología. Instructor. Hospital Pediátrico Octavio de
2
Especialista de Primer Grado en Imagenología. Instructor. Hospital Pediátrico
Octavio de
3
Especialista
de Primer Grado Neurocirugía.
Instructor. Hospital Pediátrico Octavio
de la Concepción y de la Pedraja Holguín
4
Especialista
de Primer Grado Neurocirugía.
Instructor. Hospital Pediátrico Octavio
de
5
Especialista
de Primer Grado en Neurología.
Instructor. Hospital Pediátrico Octavio de
RESUMEN
Se presentó un enfermo que padece el síndrome de Crouzon (rara
entidad heredada en forma autonómica dominante) el cual se diagnosticó mediante hallazgos clínicos confirmados por
estudios imagenológicos de una
tomografía axial computarizada realizada en agosto de 2009. Se planificó y
ejecutó la craniectomía con éxito. La evolución clínica del paciente fue
satisfactoria.
Palabras clave: síndrome de
Crouzon, tomografía
axial computarizada, tratamiento quirúrgico,
craniectomía
ABSTRACT
A patient with
a rare hereditary disease, called Crouzon´s Syndrome was studied. The clinical
diagnosis was confirmed through computer axial tomography in August
Key
words: Crouzon´s Syndrome, computer axial tomography,
surgical treatment, craniectomy.
INTRODUCCIÓN
La disostosis craneofacial congénita se conoce también
como síndrome de
Crouzon. Dicha enfermedad se caracteriza por una dismorfia craneofacial, con
cierre precoz de todas las suturas craneales acompañado de hipertensión
endocraneana, dolicocefalia,
trigonocefalia, nariz en pico de
loro, maxilar superior pequeño e inferior más grande que lo normal y exoftalmia
bilateral. Este síndrome es un trastorno genético causado por mutaciones en el
gen del receptor del factor de crecimiento de los fibroblastos tipos 2 y 3
(FGFR2 y FGFR3).
El objetivo del
trabajo fue presentar un caso diagnosticado en el Hospital Provincial
Pediátrico Universitario Octavio de
PRESENTACIÓN DE
CASO
Se presenta, con consentimiento informado, un paciente
masculino de dos años de edad con antecedentes de salud aparente, fue traído a la Consulta de
Neurología en agosto de 2009, porque el perímetro cefálico no aumentaba de
forma suficiente y presentaba una configuración craneofacial anómala.
Se le realizaron radiografías simples de cráneo en proyecciones
frontal, lateral y Town y luego se procedió a estudiar con tomografía axial
computarizada. Se realizaron cortes simples de cráneo de
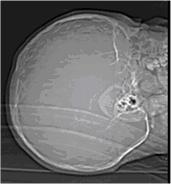
Figura 1. Topograma en Proyección
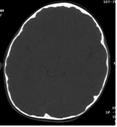

Figura 2. Corte axial tomográfico con Figura 3. Reconstrucción
ventana ósea
tomográfica
tridimensional
DISCUSIÓN
En 1912 Crouzon describió en una madre y su hijo el síndrome hereditario de
la disostosis craneofacial y define la tríada de deformidades del calvario,
faciales y exoftalmos1. Este síndrome tiene causa genética y se hereda en
forma autosómica dominante. Su origen radica en mutaciones de genes que se
relacionan con el desarrollo de las extremidades; el del receptor del factor de
crecimiento de fibroblastos tipo 2 (FGFR2) o menos comúnmente de los genes del
(FGFR3)4. Modificaciones
específicas en esos receptores son la causa de la unión precoz de los huesos
craneales.
Ese genotipo determina la manifestación fenotípica de cierre prematuro de
las suturas craneales (craneosinostosis). Otras de las anomalías acompañantes
incluyen el hipertelorismo, exoftalmos, estrabismo, maxilar superior pequeño,
prognatismo y otras menos constantes.
Su prevalencia es de un caso por sesenta mil habitantes y es el responsable
de aproximadamente el 4,8% de las craneosinostosis2.
Como complicación fundamental se encuentra la atrofia del nervio óptico por el
incremento de la presión intracraneal. El diagnóstico de este síndrome se
realiza a través de la clínica, los estudios imagenológicos y genéticos.
No se ha descrito ningún tratamiento curativo para esta
enfermedad, pero varias de las dificultades funcionales más apremiantes si
pueden ser tratadas. Dentro de los métodos quirúrgicos existe la craniectomía
(extirpación y reemplazo de porciones del hueso craneal); que realizada en los
primeros años de vida puede prevenir el daño cerebral compresivo
y mantener la forma craneal menos
deformada posible 3, 4.
También pueden operarse el exoftalmos, el prognatismo y
el paladar hendido, cuyos resultados son de éxito variable. Otras medidas
terapéuticas incluyen los métodos ortodóncicos y el tratamiento de las
complicaciones clínicas, oftalmológicas y auditivas5, 6,7. Estos
pacientes pueden requerir educación especial si presentan algún grado de
retardo mental.
En este enfermo la sospecha clínica del raro síndrome;
acentuada porque su madre presentaba dismorfia facial, evidencia de un patrón
hereditario; se confirmó con el estudio imagenológico tomográfico. La
definición anatómica precisa de las anormalidades craneales permitió planificar y ejecutar la
craniectomía con éxito. Después de realizada ésta, el paciente tuvo una
evolución acorde con los beneficios esperados de esa técnica.
REFERENCIAS BIBLIOGRÁFICAS
1. Ahmed
I, Afzal A. Diagnosis and evaluation of Crouzon syndrome. J Coll Physicians Surg Pak 2009;
19(5):318-20.
2.
Cohen MM Jr. Perspectives on
craniosynostosis. Am J Med Genet 2005;
136:313-326.
3.
Posnick J C, Ruiz RL. The craniofacial disostosis
syndromes: Current surgical thinking and future. Cleft Palate Craniofac J 2000; 37:433.
- Craniosynostosis syndromes (FGFR-related). GeneReviews
at GeneTests, GeneClinics: Medical Genetics Information Resource [database
online]. University of Washington, Seattle, 1997-2002.
<http://www.genetests.org>
- Colosimo C, Tartaro A, Cama A, Tortori-Donati P. The
craniosynostoses. In: Tortori-Donati P, Rossi A, Biancheri R (eds). Berlin
Springer: Pediatric Neuroradiology 2005: p. 1289–1315.
- Persing J A, Jane J A. Craniosynostosis. 4th
Ed.In: Youmans J R. Neurological
surgery. Philadelphia: W B Saunders, 1996.
- Crouzon syndrome. Cleft Palate Foundation
Publications [Artículo en línea] <http://www.cleftline.org/publications/cruozon.htm> [Consulta:
16 de sept. 2005].
Correspondencia: Dra. Janice Morales Rodríguez,
Ave Cajigal # 567 entre Cervantes y Fomento Holguín. Teléfono: 465469 Email:
jmrodriguez@hpuh.hlg.sld.cu