Presentación de casos
Facultad de Ciencias Médicas “Mariana Grajales Coello”. Holguín.
Heterogeneidad clínica-familiar en tres pacientes con retinosis pigmentaria.
Family- Clinical Heterogeneity in Three Patients with Retinitis Pigmentosa.
Elena Caridad Díaz Santos1, Surina Sera Velázquez2, Liliana María Batista Hernández3, Marta Milian Reyes4, Ileana Pupo Herrera5.
1. Especialista de Segundo Grado en Oftalmología. Master en Ciencias. Profesor Instructor. Hospital Docente V. I. Lenin. Holguín.
2. Especialista de Primer Grado en Oftalmología. Profesor Instructor. Hospital Doc V.I.Lenin. Holguín.
3. Especialista de Primer Grado en Oftalmología. Profesor Asistente. Hospital Doc. V.I.Lenin. Holguín.
4. Especialista de Primer Grado en Oftalmología. Profesor Asistente. Hospital Doc. V.I.Lenin. Holguín.
5. Master en Ciencias. Licenciada en Sociología. Profesor Asistente. Hospital Doc. V.I.Lenin. Holguín.
RESUMEN
Se presentaron tres hermanos con diagnóstico positivo de retinosis pigmentaria, cuyos cuadros clínicos y evolución clínica son diferentes. La paciente femenina de 12 ańos (cuadro clínico1), es remitida del hospital pediátrico al centro de retinosis con diagnóstico presuntivo de retinosis pigmentaria el cuál se confirma, las manifestaciones fondoscopicas son atípicas, con una evolución severa. La paciente femenina de 11 ańos (cuadro clínico 2), fue pesquisada a partir del diagnóstico de su hermana, las manifestaciones fondoscopicas eran completamente típicas, encontrándose en los estadios iniciales de la enfermedad, con una buena evolución. Paciente masculino de ocho ańos de edad, con miopía moderada(cuadro clínico 3), fue pesquisado a partir de su hermana mayor enferma, las manifestaciones fondoscopicas de su miopía encubrían el diagnóstico de la retinosis con una evolución moderada de ambas enfermedades. Se definió en la familia un patrón de transmisión hereditario autosómico recesivo. En el análisis de las manifestaciones oftalmológicas de estos tres hermanos, se evidencia la heterogeneidad clínica intrafamiliar de esta enfermedad.
Palabras clave: retinosis pigmentaria, heterogeneidad clínica.
ABSTRACT
Three patients (siblings) with retinitis pigmentosa, whoose clinical manifestations and evolution were different were attended at Retinitis Center.The first case was a 12-year old female patient, who came from Pediatric Hospital with presumptive diagnosis of the disease and severe evolution . The second patient was 11 years old and was in the first stages of the disease with a good evolution . The third patient was 8 years old with moderate myopia and the diagnosis of this illness was hidden with moderate evolution. Both patients the second and the third one were included in the investigation due to the diagnosis on their sister. A clinical heterogeneity was evidenced through oftalmological analysis.
Key words: retinitis pigmentosa, clinical heterogeneity.
INTRODUCCIÓN
La retinosis pigmentaria es una enfermedad ocular que produce una grave disminución de la capacidad visual, y que en muchos enfermos conduce a la ceguera. Aunque se nace con la enfermedad, es raro que se manifieste antes de la adolescencia, y prácticamente siempre lo hace de forma insidiosa, de tal modo que el enfermo no es consciente de su dolencia hasta que ésta se encuentra en fases muy avanzadas.
Es muy importante saber que no todas las retinosis pigmentarias son iguales ni conducen a la misma pérdida de visión. Es un trastorno que puede ser causado por muchos defectos genéticos. La forma clínica típica es la más frecuente en Cuba, con un 96% de incidencia en pacientes enfermos 1.
Los fotorreceptores que más se dańan en el período inicial de esta enfermedad son los bastones, hecho que aclara el por qué los enfermos mencionan como "primeros síntomas" los siguientes: una mala visión al pasar de un sitio con buena iluminación a otro con iluminación deficiente; tropiezos con los objetos que les rodean, debido a que no tienen visión periférica (indirecta), lo cual está determinado por la reducción del campo visual, que se conoce con el nombre de reducción concéntrica, visión tubular, visión en túnel o en cańón de escopeta.
Por el contrario, si la forma clínica de presentación es una de las formas atípica que en su período inicial dańa a los fotorreceptores llamados conos, afección que tiene una incidencia de un 4% en la población cubana, entonces los síntomas iniciales son: visión diurna y visión central malas; dificultad en la identificación de los colores (visión cromática alterada); alteración del campo visual en su parte central ; dificultad en la identificación; problemas con los cambios de relieves (peldańos de escaleras, hoyos) y suele ser frecuente la fotofobia, o sea, el malestar a la luz intensa.
Si se realiza un breve análisis de las manifestaciones clínicas-oftalmológicas de la retinosis pigmentaria, a las que ya hemos hecho referencia, se comprende fácilmente su "heterogeneidad clínica"2. Si a ello agregamos que en relación con la herencia, esta enfermedad tiene todos los tipos de la herencia mendeliana, y que con los avances de los estudios en la Genética Molecular es posible identificar nuevas mutaciones en los dańos al cromosoma, se comprende fácilmente por qué decimos "heterogeneidad genética"3,4.
La interpretación de los síntomas en los nińos muchas veces se hace más difícil ya que los más pequeńos no saben a ciencia cierta expresar lo que realmente sienten, en ocasiones encontramos cambios de conducta como: no jugar de noche, acostarse temprano, miedo a la oscuridad, cierto aislamiento que en ocasiones es mal interpretado como alteraciones puramente en la esfera psíquica y no condicionadas por limitaciones orgánica como sucede realmente, si a esto le adicionamos que las manifestaciones clínicas en sus inicios son escasas y en muchos pacientes la forma clínica de manifestación es la forma atípica, pues nos lleva a un diagnóstico tardío, con serias consecuencias 5,6.
Se considera el paradigma de la heterogeneidad genética como la más común dentro de las retinopatías de origen genético, presentándose con todas las formas de herencia monogénicas posibles (autosómica dominante y recesiva y ligada al cromosoma X), y siguiendo mecanismos de herencia no tradicional (herencia mitocondrial, digénica, disomía uniparental y mosaicismo germinal). Además puede aparecer de forma aislada o asociada a otros síndromes genéticos 7, 8,9.
Se plantea que más de 100 genes, en alrededor de 70 loci, están implicados en la producción de la RP 10.
Por lo que consideramos que todos los esfuerzos que se realicen para lograr el diagnóstico asintomático de una enfermedad tan invalidante como la que tratamos es poco en la lucha contra la ceguera en la cual nos sentimos partícipes como Especialista de Oftalmología por lo que nos motivamos a la realización de este estudio.
PRESENTACIÓN DE LOS PACIENTES
Se presentan tres hermanos de 8, 11 y 12 ańos, con diagnóstico positivo de retinosis pigmentaria, atendidos en el Centro Provincial de Retinosis, desde marzo de 1992, cuyos cuadros clínicos son diferentes y con evoluciones clínicas distintas., a los que se les dio seguimiento por 16 ańos.
Paciente 1:
Adolescente femenina de 12 ańos, es remitida del Hospital Pediátrico al Centro de Retinosis con diagnóstico presuntivo de retinosis pigmentaria, refería síntomas desde los ocho ańos, las manifestaciones fondoscopicas atípicas.
Se le realizó un estudio oftalmológico completo: agudeza visual (AV), biomicroscopia, tonometría, motilidad ocular, reflejos pupilares, fondo de ojo, electrorretinograma standardizado y estudio campimétrico.
La AV era de 0,6 OD y de 0,5 en OI, al momento del diagnóstico, el ERG no registrable, campo visual a veinte grados centrales, el examen oftalmológico arrojó, una imagen fondóscopica atípica, por la ausencias de pigmentos.
Se le realiza un estudio completo por el grupo multidisciplinario de atención médica especializada a estos paciente para definir si estábamos ante la presencia de una Retinosis sistémica o no sistémica, encontrándose sólo manifestaciones a nivel ocular. Se le da seguimiento por consulta cada tres meses, evaluando los parámetros antes seńalados, a los cinco ańos de evolución la agudeza visual era de 0,5 OD y 0,4 OI, con una pérdida de cinco grados en su campo visual central, desde entonces comienza una caída rápida de su agudeza y campo visual a pesar de haber recibido tratamientos médico y quirúrgico, en los momentos actuales tiene 0,1 OD y 0,05 OI, campo visual -5 grado, Fo: sin pigmentos.
Paciente 2:
Adolescente, femenina de 11 ańos, refería síntomas desde hace sólo un ańo, fue pesquisada a partir del diagnóstico de su hermana, las manifestaciones fondoscopicas eran completamente típicas, se encontró en los estadios iniciales de la enfermedad.
Se realizó un estudio oftalmológico completo: agudeza visual (AV), biomicroscopia, tonometría, motilidad ocular, reflejos pupilares, fondo de ojo, electrorretinograma estandardizado y estudio campimétrico.
La AV era de 1,0 OD y de 1,0 en OI, al momento del diagnóstico, el ERG no registrable, campo visual a más 30 grados centrales, ventanas periféricas amplias, el examen oftalmológico arrojó, una imagen fondóscopica típica, con pigmentos escasos en periferia media.
Se le realiza un estudio completo por el grupo multidisciplinario de atención médica especializada a este paciente para definir si estábamos ante la presencia de una retinosis sistémica o no sistémica, encontrándose sólo manifestaciones a nivel ocular.
Se le da seguimiento por consulta cada seis meses, para evaluar los parámetros antes seńalados, a los cinco ańos de evolución la agudeza visual se mantenía estable, pero con una pérdida de cinco grado en su campo visual central.
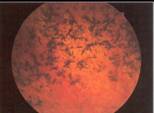
Fo: aumento de pigmentos en periferia media, que se mantuvo estable en los siguientes cinco ańos, en los momentos actuales tiene 0,8 OD y 1,0 dif en OI, campo visual veinticinco grados centrales, con área visual periférica amplia en AO.
Fo: pigmentos abundantes en periferia media .
|
Fig. 1 |

Paciente 3:
Nińo de 8 ańos de edad, de sexo masculino, con miopía moderada, se atendía en Consulta de Oftalmología con diagnóstico de una miopía moderada, fue pesquisado a partir de su hermana mayor enferma, las manifestaciones fondoscopicas de su miopía encubrían el diagnóstico de la retinosis.
Se realizó un estudio oftalmológico completo: agudeza visual (AV), biomicroscopia, tonometria, motilidad ocular, reflejos pupilares, fondo de ojo, electrorretinograma estandardizado y estudio campimétrico.
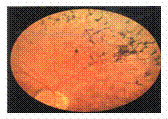
La AV era de 0,8 OD y de 0,7 en OI, con corrección óptica, al momento del diagnóstico, el ERG no registrable, campo visual a veinticinco grados centrales, ventanas periféricas amplias. El examen oftalmológico arrojó, una imagen fondóscopica característica de su ametropía, con pigmentos abundantes en periferia media.
|
Fig. 2 |
Fig. 3 |
Se le realiza un estudio completo por el grupo multidisciplinario de atención médica especializada a este paciente para definir si estábamos ante la presencia de una retinosis sistémica o no sistémica, se encontró sólo manifestaciones a nivel ocular.
Se le da seguimiento por consulta cada seis meses, para evaluar los parámetros antes seńalados, a los cinco ańos de evolución la agudeza visual se mantenía estable, así como su campo visual; a los diez ańos de seguimiento ocurrió una caída de agudeza visual de una líneas del optotipo en su OD y dos líneas en su OI, no varió su campo visual, en los momentos actuales tiene 0,7 OD y 0,5 en OI, con progreso de dos dioptrías y su campo visual veinticinco grados centrales, con área visual periférica amplia en AO.
Fo: pigmentos abundantes en periferia media.
DISCUSIÓN
La clasificación cubana de la retinosis pigmentaria abarca formas clínicas de presentación, estadio clínico, tipo de transmisión hereditaria y edad de debut de la enfermedad 2.
En nuestro estudio se puede definir que las características clínicas de la enfermedad en la primera paciente fue una forma atípica de presentación, la llamada retinosis pigmentaria sin pigmentos, ya que estos no aparecieron en la evolución durante 16 ańos, con una evolución natural de la enfermedad, en la cual fue imposible variar su avance agresivo.
En los otros dos hermanos aunque con condiciones diferentes, la evolución ha sido más benigna, con una forma clínica típica, que en el paciente tres se ve influenciada por la asociación de miopía, por lo que existió gran variabilidad clínica intra-familiar. Se definió un patrón de transmisión hereditario autosómico recesivo en esta familia. En el análisis de las manifestaciones oftalmológicas y evolutivas de la enfermedad en estos tres hermanos, se evidencia la heterogeneidad clínica de la misma.
BIBLIOGRAFÍA